Our lab at the Lewis-Sigler Institute for Integrative Genomics and the Department of Computer Science at Princeton University uses applied statistics, machine learning and efficient algorithms to address fundamental problems in biology and medicine by integrative analysis of multi-dimensional data.

Research
We develop computational methods for design and analysis of high-throughput functional genomic assays and perturbations, with a focus on multi-modal single-cell, spatial and genome editing technologies. We apply these methods to study regulatory genomics of cell function and cell-cell interactions in vivo, with a focus on immunology and cancer.
Examples of our past and ongoing work are presented below.
CD8 T cells form the central component of the adaptive immune system and are essential in defense against viral and bacterial infections and in tumor immunity. Better molecular characterization of CD8 T cells in different contexts is fundamentally important and can lead to improved clinical results in cancer immunotherapies, infectious diseases, autoimmunity. We performed an extensive genomic, single-cell, and transcription factor analysis of CD8 T cell functional and dysfunctional states. We now continue studying regulatory mechanisms and cell-cell interactions governing T cell activation and functional commitment across immune challenges in mouse and human using single-cell and spatial multi-omics and direct cell-cell interaction profiling. We are also expanding these approaches to other immune cell types.

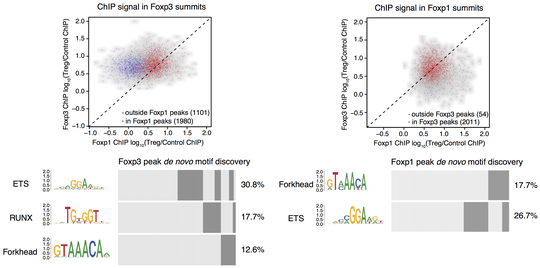
Regulatory T (Treg) cells are critical for tolerance to self-antigens and preventing autoimmunity. Their differentiation and function are controlled by transcription factor Foxp3, but mechanistic understanding of Foxp3 role remains elusive. Using functional genomic analysis, we explored Foxp3 function and its interaction with a highly expressed closely related factor Foxp1. We are now studying the role of Foxp3 and other factors in chromatin organization and gene expression regulation in functionally heterogeneous Treg cells across tissues.
Programmable genome editing using CRISPR has tremendously advanced life sciences. To facilitate the use of this technology, especially in the noncoding genome and for batch screens, we developed GuideScan, a fully customizable CRISPR guide RNA (gRNA) design tool. We recently developed GuideScan2 with substantially expanded and new functionality for memory-efficient, parallelizable construction of high-specificity gRNA databases and user-friendly gRNA design in custom genomes. We now continue working on tools for design and analysis of CRISPR-based genome perturbations and their combinations with single-cell functional genomic assays.

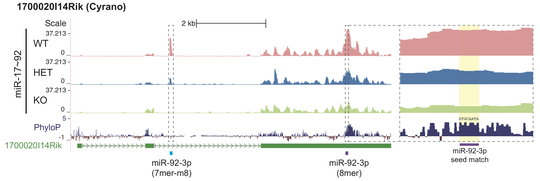
Cross-linking immunoprecipitation followed by sequencing (CLIP-seq) is a family of methods for profiling sites of protein binding to RNA. In particular, CLIP has been used to identify targets of microRNAs, small non-coding RNA molecules that bind to a protein Ago2 and thus regulate gene expression post-transcriptionally. We developed a new algorithm CLIPanalyze for analysis of such data and used it for comprehensive analysis of microRNA targets in vivo in mouse embryonic stem cells, developing embryos, adult tissues and multiple cancer models. We now continue development and application of methods for studies of post-transcriptional regulation.
News
People
Principal Investigator
Yuri Pritykin
Assistant Professor of Computer Science and Genomics
Current Lab Members
Tamjeed Azad
PhD student
Vineet Bansal
Research Software Engineer
Yanjin Chen
PhD student (co-advised with Britt Adamson)
Jonathan Mitchel
Postdoctoral Research Associate
Wenfan Ke
Postdoctoral Research Associate
Tim Kosfeld
PhD student
Dillon Lue
PhD student
Alexander Ostrin
Research Specialist
Amanda Sun
PhD student
Ian Traniello
Lewis-Sigler Scholar (co-advised with Sarah Kocher)
Brendan Wang
Research Specialist
Yingying Zhang
Lewis-Sigler Scholar (co-advised with Mona Singh)
Zhiyue Zhang
PhD student (co-advised with Lydia Lynch)
Alumni
Dimitar Chakarov
Undergraduate Research Assistant
Gabriel Dolsten
PhD student
Yujie Qiao
Research Specialist
Zhengqiao Zhao
Postdoctoral Research Associate
Adina Scheinfeld
Research Specialist
Sarah Walker
PhD student
Selected publications
Publications
Teaching
- Fall 2025: COS 126 Computer Science: An Interdisciplinary Approach
- Spring 2025: QCB 311 / MOL 311 / COS 311 / EEB 311 Genomics
- Fall 2024: COS IW 04 / 05 Computation and Machine Learning in Life Sciences and Biomedicine
- Spring 2024: QCB 311 / MOL 311 / COS 311 Genomics
- Fall 2023: COS 126 Computer Science: An Interdisciplinary Approach (Biomedical precept)
- Spring 2023: QCB 311 / COS 311 Genomics
- Fall 2022: COS IW 06 Computation and Machine Learning in Life Sciences and Biomedicine
- Fall 2021: COS 597D Advanced Computational Genomics
Join us
We are actively looking for outstanding scientists at all levels (postdocs, graduate and undergraduate students, staff scientists) across disciplines (computational biology, computer science, machine learning, applied mathematics, immunology, molecular biology) to join our team. Please contact us if interested.
Postdoctoral positions in computational biology are available. We seek candidates with computational, bioinformatics, machine learning, statistics, data science and/or other quantitative backgrounds who are enthusiastic about bringing their expertise to address fundamental problems in biology and medicine using cutting-edge technologies. A successful candidate will have an opportunity to lead and contribute to a range of exciting collaborative projects, and develop new projects. Particularly, positions focused on regulatory genomics and/or immunology, as well as genome editing, are available. Please contact us to learn more.
Prospective PhD students interested in working with us are encouraged to apply to Computer Science, Quantitative and Computational Biology, Molecular Biology or Bioengineering graduate programs. We are also accepting students via the CS Master of Science in Engineering program.
Current Princeton graduate and undergraduate students interested in working with us are encouraged to contact us directly.
Contact
- pritykin@princeton.edu
- 245 Carl Icahn Lab, Lewis-Sigler Institute for Integrative Genomics, South Drive, Princeton University, Princeton, NJ 08540